Abstract:
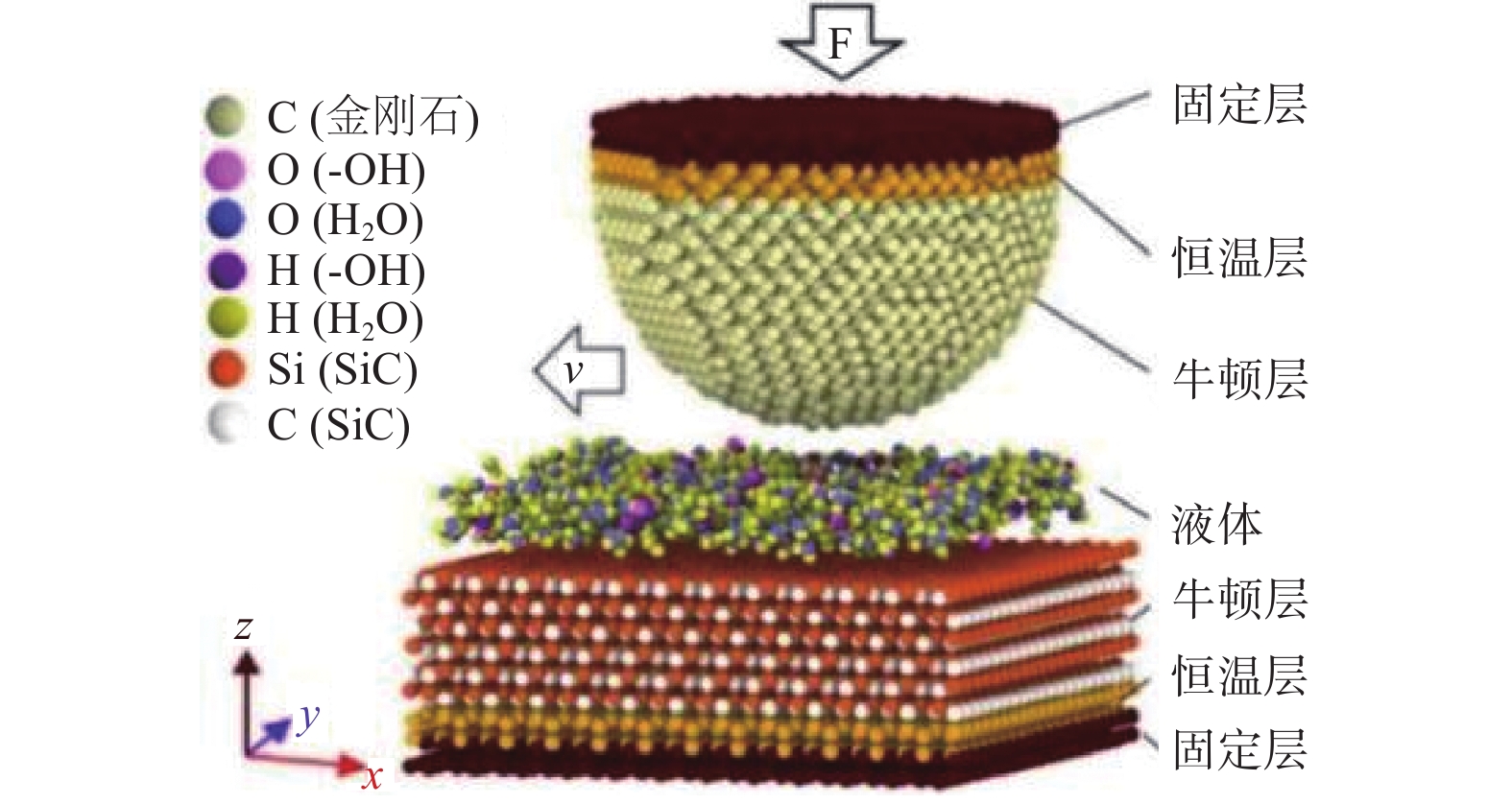
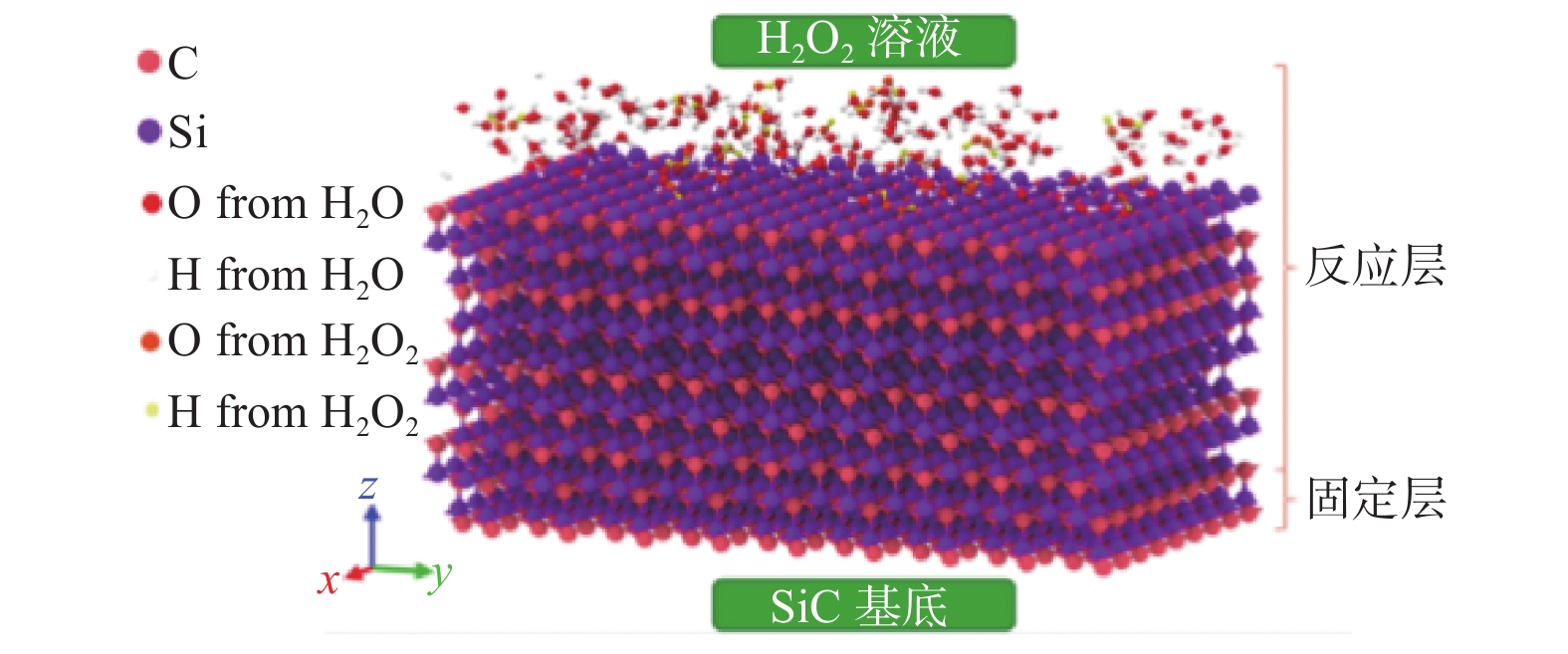
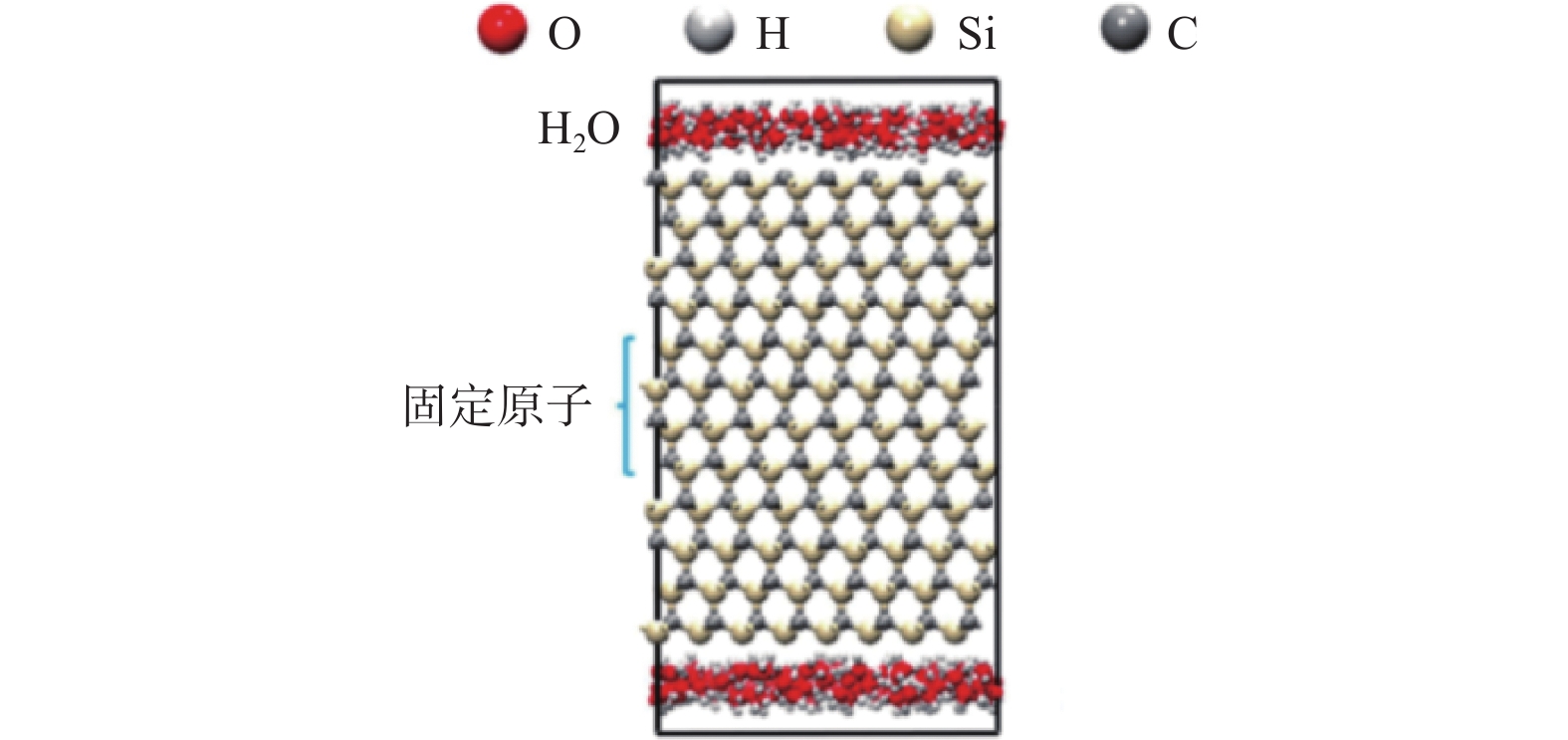
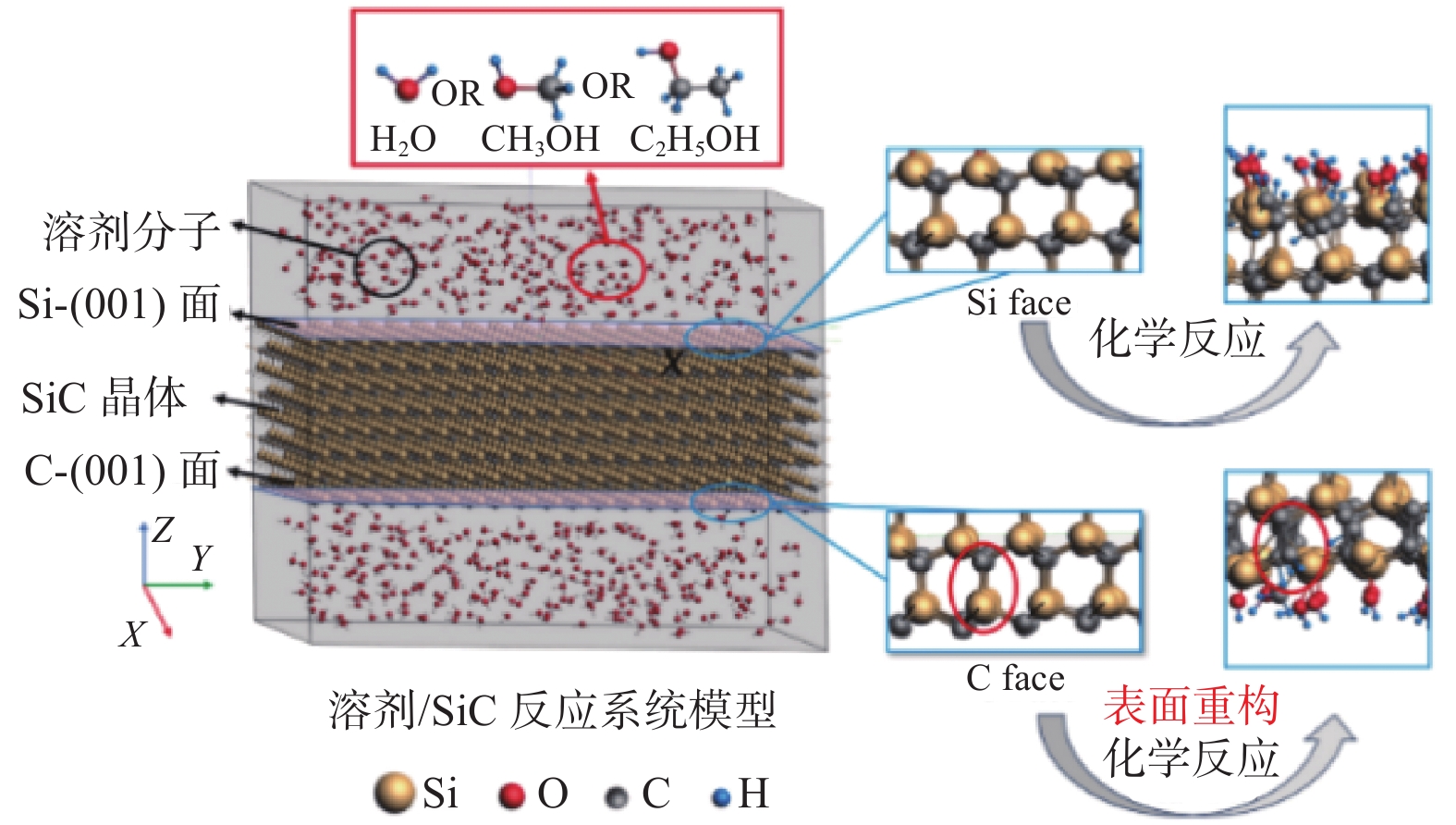
Significance Silicon carbide (SiC), as a representative material of third-generation semiconductors, holds vast potential for applications in microelectronics, optoelectronics, aerospace, and energy. However, its high hardness and chemical stability pose significant challenges for processing. Chemical mechanical polishing (CMP) is a crucial technology for planarizing SiC substrates. It can effectively remove the damaged layer and impurities on the wafer surface, achieve a high degree of planarization, thereby enhance the performance and reliability of SiC devices. Extensive research has been conducted on CMP processes, yet the mechanisms of interaction and synergy among abrasives, solution media, and SiC surfaces remain unclear. Molecular dynamics (MD) simulation, based on Newton's laws of motion and the principles of quantum mechanics, is a simulation method used to reveal the interactions between the microscopic structure and properties of matter. It is currently widely applied in the study of SiC surface removal mechanisms. By simulating the scratching behavior of abrasives on SiC surfaces, changes in material morphology, crystal structure, temperature, cutting force, and potential energy can be observed, thereby providing deeper insights into polishing mechanisms. This in-depth understanding of polishing mechanisms aids in optimizing polishing process parameters, improving polishing efficiency, and surface quality. Meanwhile, during the SiC CMP process, certain components in the polishing solution interact with the SiC surface, potentially involving a series of chemical reactions. MD simulation can reveal the detailed mechanisms of these chemical reactions, including the reaction pathways, reaction rates, and reaction products, thereby facilitating a deeper understanding of the material removal mechanism during the polishing process and providing a theoretical basis for optimizing polishing processes. Progress The article first analyzes the potential functions commonly used in MD simulations for SiC precision polishing and summarizes their application fields. It then integrates and analyzes existing MD simulation studies on SiC CMP. MD simulations for SiC substrate precision polishing are mainly classified into three categories: SiC material properties, abrasive grinding, and SiC surface chemical reactions. The Tersoff potential function has been widely applied in the preparation and properties of SiC materials, demonstrating excellent simulation results. It has become the most popular potential function for MD simulations of SiC materials. The Tersoff / ZBL potential function enhances the Tersoff potential function by incorporating the ZBL potential, thus adding short-range interactions and providing a more accurate description of short-range atomic collisions. The ABOP potential function, based on the Tersoff potential function, allows for the breaking of chemical bonds, making it more suitable for simulating wear behavior. The Vashishta potential function is well-suited for accurately simulating the deformation of ionic and covalent bonds in 3C-SiC, including bending and stretching. It is widely used in simulations involving impact behavior and nanoindentation of SiC. The advantage of the ReaxFF lies in its ability to simulate the formation and breaking of bonds during chemical reactions, making it suitable for simulating chemical reactions, adsorption, and other phenomena on SiC surfaces. Conclusions and Prospects Currently, many aspects of the CMP mechanism of SiC materials remain unclear. MD simulations can be utilized to study the interaction mechanisms between liquids, oxides, and surfaces during CMP, such as charge transfer and surface adsorption. Most research has focused on the mechanical interactions between abrasives and SiC surfaces, with relatively little attention paid to chemical reaction mechanisms. Future research will emphasize using the ReaxFF through MD simulations to study the reaction mechanism of SiC under various conditions, developing more potential functions to accommodate different polishing conditions, and establishing comprehensive models to consider the impact of multiple factors on surface interactions. During MD simulations of SiC oxidation mechanisms, different potential functions have distinct application fields. Although the ReaxFF reactive force field can effectively simulate SiC surface oxidation reactions, using the Tersoff potential function to simulate the interaction between SiC and abrasives is more reasonable. Due to the high modeling proficiency required to establish mixed potential function models combining the ReaxFF reactive force field with other potential functions, researchers often adopt the ReaxFF single intermolecular potential for calculations. If oxidation reactions and abrasive grinding occur simultaneously during the calculation process, it may not accurately describe the SiC surface interaction mechanisms. Therefore, combining the ReaxFF with other potential functions to achieve MD simulation of chemical mechanical polishing under the combined action of multiple factors will be a direction for future research.

 Abstract
Abstract HTML
HTML PDF 8603KB
PDF 8603KB Cited By
Cited By







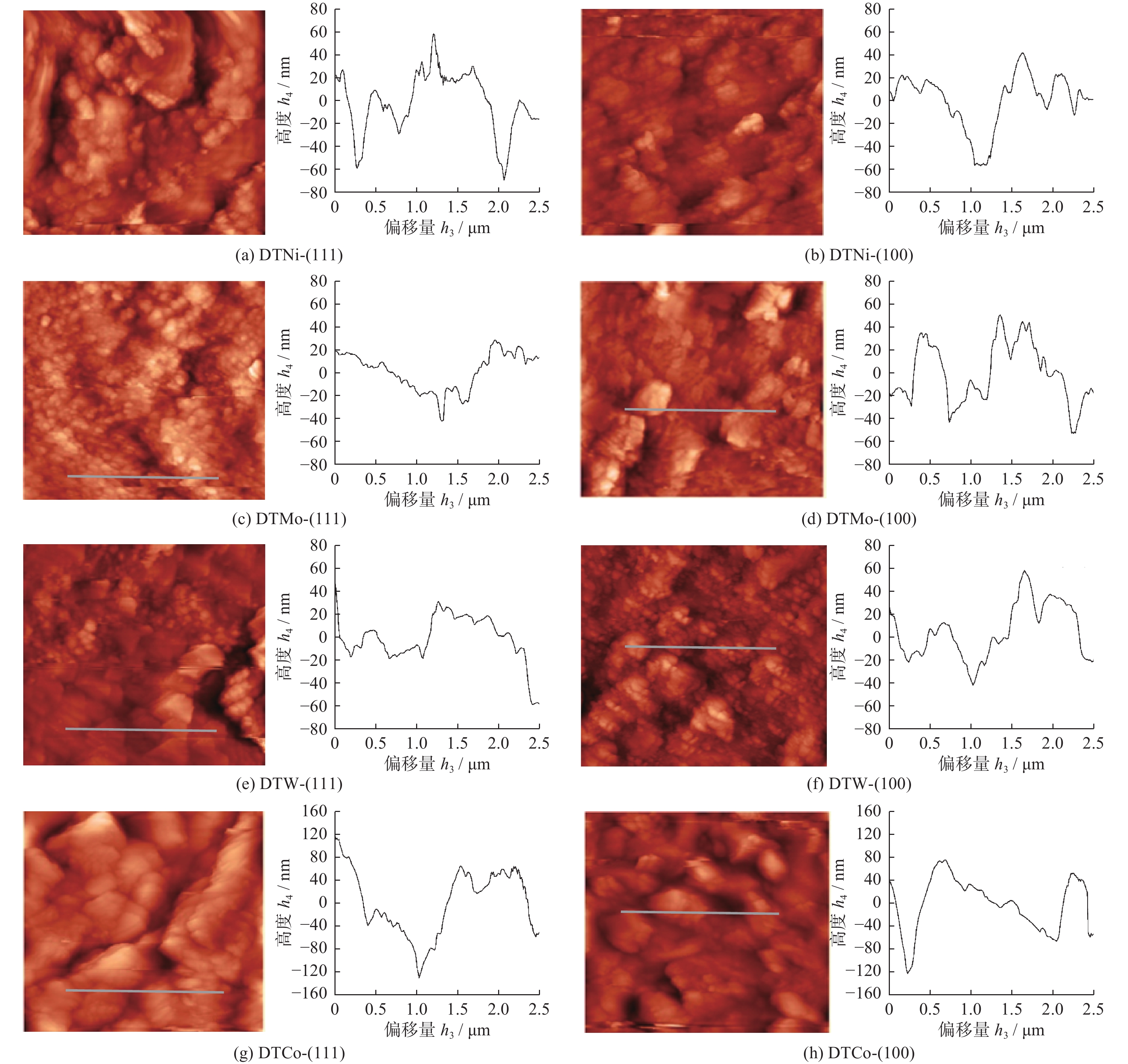
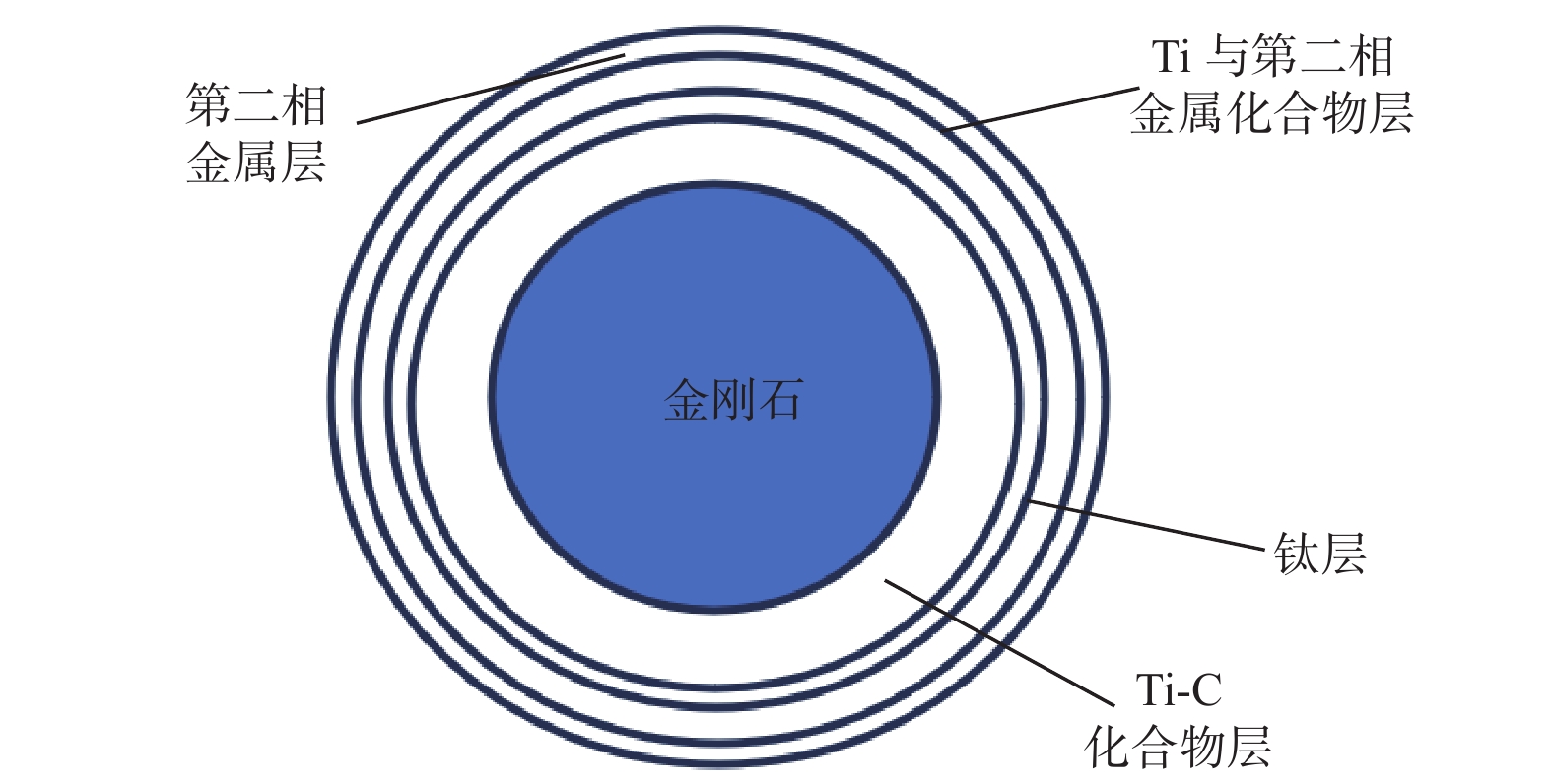



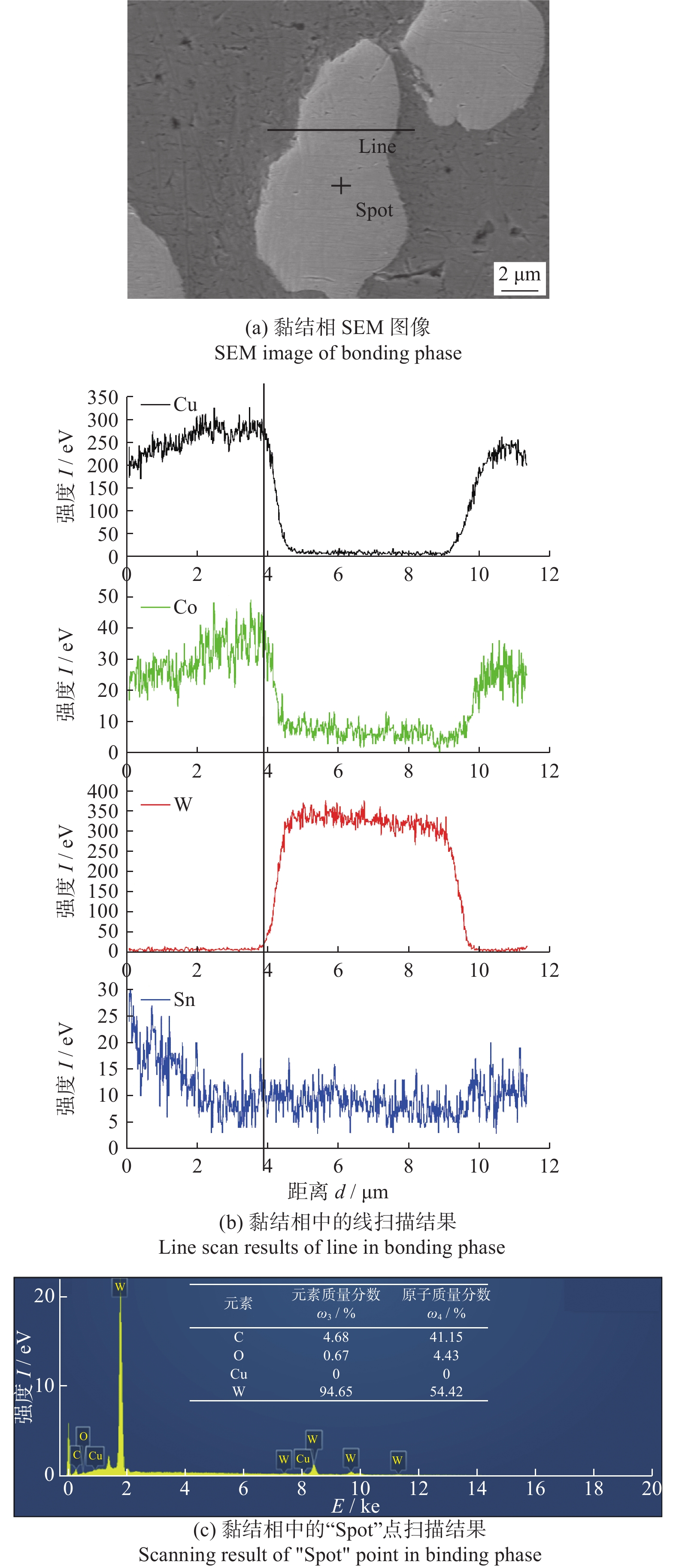
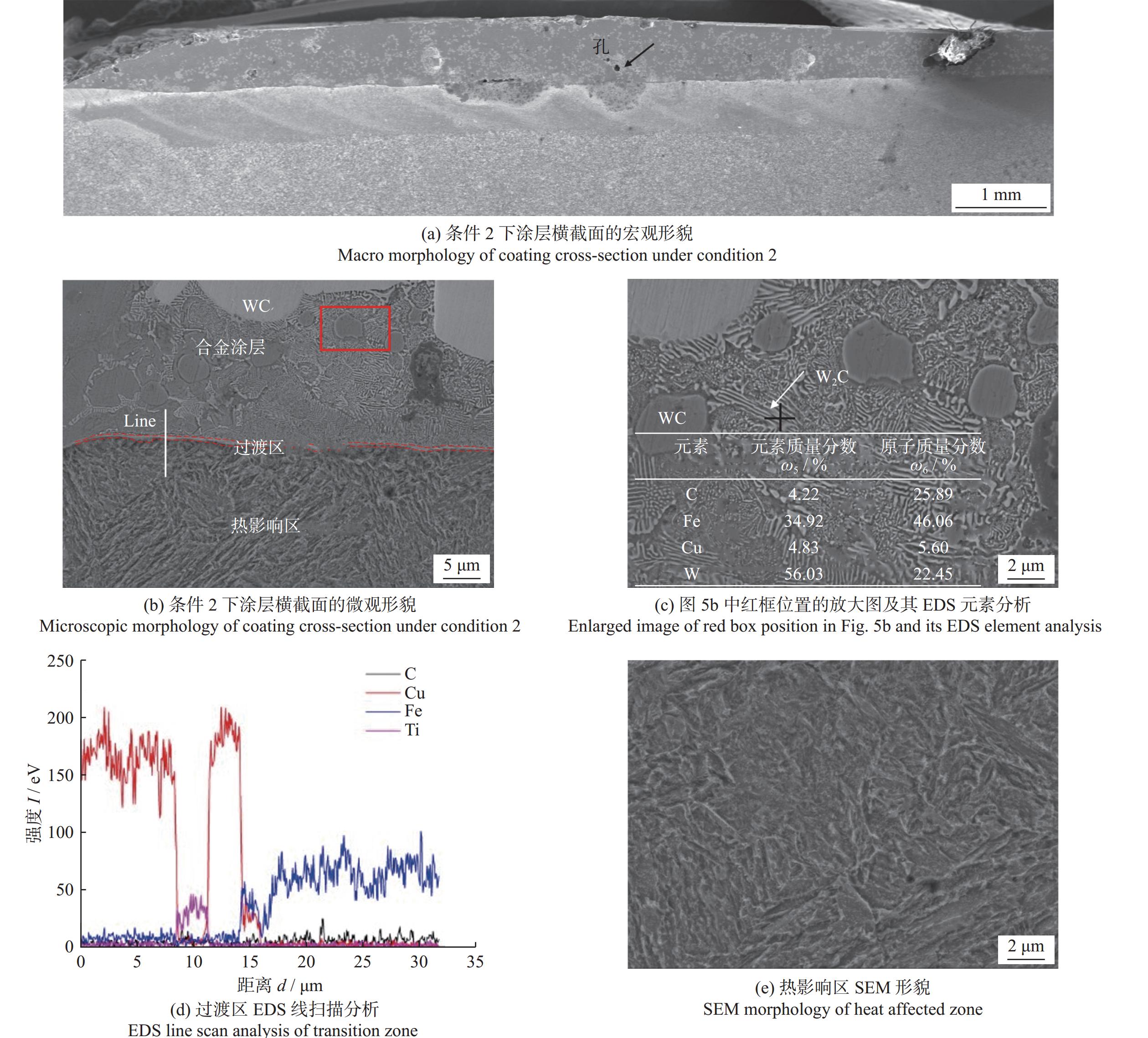
































 Email Alerts
Email Alerts RSS
RSS